Centre de référence des déficiences intellectuelles et polyhandicaps de causes rares
![]() Le site constitutif de l’hôpital universitaire Necker-Enfants malades a développé une expertise dans le soin, l’enseignement et la recherche dans le polyhandicap lié à des encéphalopathies développementales telles que le syndrome de Rett et les syndromes génétiques apparentés (CDKL5, FOXG1, syndrome d’Aicardi, syndrome d’Angelman) et les malformations cérébrales.
Le site constitutif de l’hôpital universitaire Necker-Enfants malades a développé une expertise dans le soin, l’enseignement et la recherche dans le polyhandicap lié à des encéphalopathies développementales telles que le syndrome de Rett et les syndromes génétiques apparentés (CDKL5, FOXG1, syndrome d’Aicardi, syndrome d’Angelman) et les malformations cérébrales.
L’équipe ![]() apporte également un soin particulier à la prise en charge pluridisciplinaire du polyhandicap, en combinant une évaluation neurologique aux évaluations gastro-entérologiques, endocrinologiques, orthopédiques et respiratoires de nos patients. De la même manière, elle développe des relations privilégiées avec le secteur médico-social afin de participer au projet de vie des patients suivis dans le centre.
apporte également un soin particulier à la prise en charge pluridisciplinaire du polyhandicap, en combinant une évaluation neurologique aux évaluations gastro-entérologiques, endocrinologiques, orthopédiques et respiratoires de nos patients. De la même manière, elle développe des relations privilégiées avec le secteur médico-social afin de participer au projet de vie des patients suivis dans le centre.
Notre centre est l’un des plus gros centres européens de prise en charge des patients atteints de syndromes de Rett et de malformations cérébrales.
Depuis la labellisation en 2017, ce centre de référence maladies rares s’appuie sur le réseau national de centres de compétence du centre de référence des déficiences intellectuelles de causes rares, répartis sur l’ensemble du territoire national y compris les DOM et TOM. Ceci permet pour les patients atteints de ces formes de polyhandicap un accès aux soins facilité et l’utilisation la plus optimale des compétences et ressources de chacun.
![]() Ce centre de référence est affilié à la filière de santé maladies rares DEFISCIENCE et au
Ce centre de référence est affilié à la filière de santé maladies rares DEFISCIENCE et au ![]() réseau européen de référence (ERN) ITHACA.
réseau européen de référence (ERN) ITHACA.
Médecin
responsable
Pr Nadia Bahi-Buisson
Pour prendre rendez-vous
Tél. 01 42 19 26 61
En cas
d’urgence

- Pathologies concernées
- Équipe de Necker
- Protocoles nationaux de diagnostic et de soins (PNDS)
- Recherche
- Enseignement
- Guides de bonne pratique et ouvrages didactiques
- Publications
- Retour d'expérience : la télémédecine pour des patients polyhandicapés
- Associations de patients
- Revue de presse
- Carte du réseau national
- Informations Covid
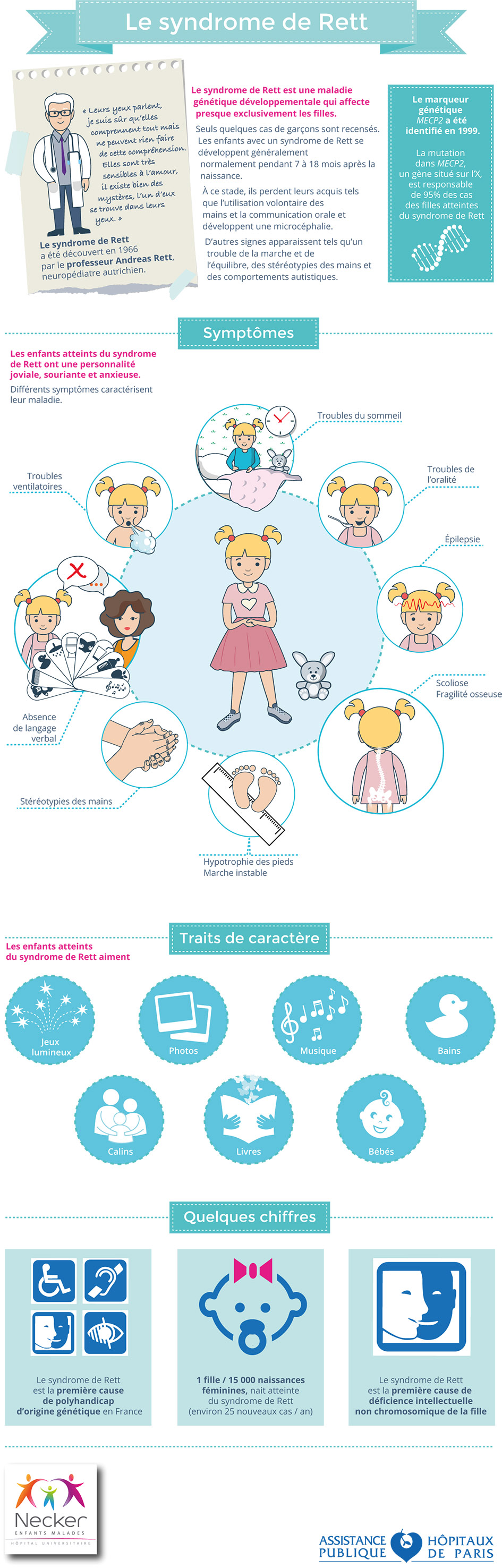
- Le syndrome de Rett typique est une maladie développementale qui affecte presque exclusivement les filles. Seuls quelques cas de garçons sont recensés. Les enfants avec un syndrome de Rett se développent généralement normalement pendant 7 à 18 mois après la naissance. À ce stade, ils perdent leurs acquis tels que l’utilisation volontaire des mains et la communication orale et développent une microcéphalie. D’autres signes apparaissent tels qu’un trouble de la marche et de l’équilibre, des stéréotypies des mains et des comportements autistiques. Le syndrome de Rett évolue vers un polyhandicap avec une durée de vie moyenne de 50 ans. La forme classique du syndrome de Rett est liée dans plus de 90% des cas à une mutation de MECP2, situé sur le chromosome X. Des résultats expérimentaux obtenus chez la souris et les neurones humains in vitro ont permis de démontrer que même les symptômes les plus sévères du syndrome de Rett pourraient être « réversés » par la réintroduction d’une forme non mutée de MecP2. Ainsi, tous les efforts de la recherche académique et industrielle sont aujourd’hui concentrés sur la recherche de ce traitement.
- Les syndromes de Rett atypiques, variants et polyhandicap associé à une microcéphalie, les syndromes FOXG1 et « FOXG1 plus », l’encéphalopathie liée aux mutations CDKL5
Un certain nombre d’encéphalopathies développementales partagent des symptômes avec le syndrome de Rett, tels que les syndromes CDKL5 ou FOXG1. De la même manière, le syndrome de Rett a des caractéristiques communes avec des anomalies du développement cérébral telles que les microcéphalies congénitales. - Syndrome d’Angelman
- Syndrome d’Aicardi
- Polyhandicap lié à des anomalies du développement du cortex cérébral (lissencéphalie, polymicrogyrie, microcéphalie/ mégalencéphalie, hétérotopies)


![]()
Dr Marie Hully, neurologie pédiatrique
Béatrice Gaschignard, coordinatrice de parcours de santé
Dr Cécile Talbotec, gastro-entérologie pédiatrique
Pr Brigitte Fauroux et Dr Alessandro Amaddeo, pneumologie pédiatrique et unité de ventilation – spécialistes du sommeil
Dr Robert Rubinsztajn, réanimation pédiatrique
Dr Hauviette Descamps, médecine physique et réadaptation
Dr Lotfi Miladi et Dr Alina Badina, orthopédie pédiatrique
Pr Dominique Bremond-Gignac, ophtalmologie pédiatrique
Dr Lisa Ouss, pédo-psychiatrie
Camille Compte, assistante sociale
Les protocoles nationaux de diagnostic et de soins (PNDS) sont des référentiels de bonne pratique portant sur les maladies rares. L’objectif d’un PNDS est d’expliciter aux professionnels concernés la prise en charge diagnostique et thérapeutique optimale et le parcours de soins d’un patient atteint d’une maladie rare donnée.
Comme le prévoit le deuxième plan national maladies rares 2011-2014, ils sont élaborés par les centres de référence et de compétence maladies rares à l’aide d’une méthode proposée par la Haute Autorité de Santé (HAS).
PNDS élaborés par le centre de référence
L’équipe du centre Rett travaille en étroite collaboration avec le groupe de recherche animé par le Pr Bahi- Buisson dans l’équipe du Dr Alessandra Pierani -équipe génétique et développement cérébral Imagine Institute INSERM UMR-1163- Paris Descartes University.
L’équipe de recherche est composée de :
- Pr Nadia Bahi-Buisson, MD-PhD, group leader
- Dr Amandine Bery, post-doctorante
- Dr Mara Cavallin, MD, doctorante 3e année – génétique du syndrome d’Aicardi et des malformations cérébrales
- Sarah Farcy, doctorante 1re année – dynéinopathies
- Camille Maillard, doctorante 1re année – projet FOXG1
Anciens étudiants :
- Nancy Vegas, MD, Master 2
- Marion Philbert, MD, Master 2
Les principaux projets financés sont les suivants :
- ANR 2016-2019 Elucidating molecular and cellular mechanisms underlying dyneinopathies
- EraNet Neuron 2015-2018 Stem cells aiding comprehension of human malformations related to cortical development
- COST Action OC-2016-1-20862 European Network on Brain Malformations NeuroMIG (COST intergovernmental framework for European Cooperation in Science and Technology, allowing the coordination of nationally-funded research on a European level)
- Fondation Maladies Rares 2016: Identification of the genetic bases of Aicardi syndrome
- Fondation Maladies Rares 2015 : Investigating Novel molecular basis for periventricular nodular heterotopia
- Rôle de FOXG1/TLE1 dans le switch prolifération-différentiation des progéniteurs neuronaux
- AFSR 2017 : Evaluation des stéréotypies et de leur prise en charge dans le syndrome de Rett
DIU neurologie pédiatrique
Paris V –Paris VII – Paris XI – Amiens – Angers –Lille – Marseille – Montpellier- Reims – Toulouse -Tours
Livre « Syndrome de Rett » à destination des médecins, parents et aidants Coordination de rédaction AFSR; ed. 2016
AICARDI_Diseases_Childhood_3rd Chapitre Malformations cérébrales
Atlas of epilepsy: chapitre Rett Syndrome
Handbook of clinical Neurology : chapitre Rett syndrome
Handbook of clinical Neurology : chapitre Cerebral Malformations
Encyclopédie Orphanet du Handicap : Le syndrome de Rett
Principes de Médecine Interne: 18ème édition : chapitre Malformations corticales
Flammarion Médecine-Sciences Neurologie Pédiatrique : chapitre : syndrome de Rett
2021
– Deep phenotyping unstructured data mining in an extensive pediatric database to unravel a common KCNA2 variant in neurodevelopmental syndromes.
Marie Hully, Tommaso Lo Barco, Anna Kaminska, Giulia Barcia, Claude Cances, Cyril Mignot, Isabelle Desguerre, Nicolas Garcelon, Edor Kabashi, Rima Nabbout
Genet Med, 2021 May, PMID: 33500571 PMCID: PMC8105164 DOI: 10.1038/s41436-020-01039-z
– Human neuropathology confirms projection neuron and interneuron defects and delayed oligodendrocyte production and maturation in FOXG1 syndrome.
Nina-Maria Wilpert, Florent Marguet, Camille Maillard, Fabien Guimiot, Jelena Martinovic, Séverine Drunat, Tania Attié-Bitach, Ferechté Razavi, Aude Tessier, Yline Capri, Annie Laquerrière, Nadia Bahi-Buisson
Eur J Med Genet, 2021 Sep, PMID: 34284163 DOI: 10.1016/j.ejmg.2021.104282
– The phenotypic spectrum of X-linked, infantile onset ALG13-related developmental and epileptic encephalopathy.
Alexandre N. Datta,Nadia Bahi-Buisson,Thierry Bienvenu,Sarah E. Buerki,Fiona Gardiner,J. Helen Cross,Bénédicte Heron,Anna Kaminska,Christian M. Korff,Anne Lepine,Gaetan Lesca,Amy McTague,Heather C. Mefford,Cyrill Mignot,Matthieu Milh,Amélie Piton,Ronit M. Pressler,Susanne Ruf,Lynette G. Sadleir,Anne de Saint Martin,Koen Van Gassen,Nienke E. Verbeek,Dorothée Ville,Nathalie Villeneuve,Pia Zacher,Ingrid E. Scheffer,Johannes R. Lemke
Epilepsia, 2021 Feb, PMID: 33410528 PMCID: PMC7898319 DOI: 10.1111/epi.16761
– Heterogeneity in defining fetal corpus callosal pathology: systematic review.
H Mahallati, A Sotiriadis, C Celestin, A E Millischer, P Sonigo, D Grevent, N O’Gorman, N Bahi-Buisson, T Attié-Bitach, Y Ville, L J Salomon
Ultrasound Obstet Gynecol, 2021 Jul, PMID: 32798278 DOI: 10.1002/uog.22179
– Basal Ganglia Dysmorphism in Patients With Aicardi Syndrome.
Silvia Masnada, Anna Pichiecchio, Manuela Formica, Filippo Arrigoni, Paola Borrelli, Patrizia Accorsi, Paolo Bonanni, Renato Borgatti 1, Bernardo Dalla Bernardina, Alberto Danieli, Francesca Darra, Nicolas Deconinck Valentina De Giorgis, Olivier Dulac, Svetlana Gataullina, Lucio Giordano, Renzo Guerrini, Francesca La Briola, Massimo Mastrangelo, Martino Montomoli, Marzia Mortilla, Elisa Osanni, Pasquale Parisi, Emilio Perucca, Lorenzo Pinelli, Romina Romaniello, Mariasavina Severino, Federico Vigevano, Aglaia Vignoli, Nadia Bahi-Buisson et al.
Neurology, 2021 Mar 2, PMID: 33277420 PMCID: PMC8055324 DOI: 10.1212/WNL.0000000000011237
– Dysregulation of the NRG1/ERBB pathway causes a developmental disorder with gastrointestinal dysmotility in humans.
Thuy-Linh Le, Louise Galmiche, Jonathan Levy, Pim Suwannarat, Debby Mei Hellebrekers, Khomgrit Morarach, Franck Boismoreau, Tom Ej Theunissen, Mathilde Lefebvre, Anna Pelet, Jelena Martinovic, Antoinette Gelot, Fabien Guimiot, Amanda Calleroz, Cyril Gitiaux, Marie Hully, Olivier Goulet, Christophe Chardot, Severine Drunat, Yline Capri, Christine Bole-Feysot et al.
J Clin Invest, 2021 Mar 15, PMID: 33497358 PMCID: PMC7954599 DOI: 10.1172/JCI145837
– Inherited glycosylphosphatidylinositol defects cause the rare Emm-negative blood phenotype and developmental disorders.
Romain Duval, Gaël Nicolas, Alexandra Willemetz, Yoshiko Murakami, Mahmoud Mikdar, Cedric Vrignaud, Hisham Megahed, Jean-Pierre Cartron, Cecile Masson, Samer Wehbi, Bérengere Koehl, Marie Hully, Karine Siquier, Nicole Chemlay, Agnes Rotig, Stanislas Lyonnet, Yves Colin, Giulia Barcia, Vincent Cantagrel, Caroline Le Van Kim, Olivier Hermine, Taroh Kinoshita, Thierry Peyrard, Slim Azouzi
Blood, 2021 Jul 1, PMID: 33763700 DOI: 10.1182/blood.2020009810
– High rate of hypomorphic variants as the cause of inherited ataxia and related diseases: study of a cohort of 366 families.
Mehdi Benkirane, Cecilia Marelli, Claire Guissart, Agathe Roubertie, Elizabeth Ollagnon, Ariane Choumert, Frédérique Fluchère, Fabienne Ory Magne, Yosra Halleb, Mathilde Renaud, Lise Larrieu, David Baux, Olivier Patat, Idriss Bousquet, Jean-Marie Ravel, Danielle Cuntz-Shadfar, Catherine Sarret, Xavier Ayrignac et al.
Genet Med, 2021 Nov, PMID: 34234304 DOI: 10.1038/s41436-021-01250-6
– Enhanced cGAS-STING-dependent interferon signaling associated with mutations in ATAD3A.
Alice Lepelley, Erika Della Mina, Erika Van Nieuwenhove, Lise Waumans, Sylvie Fraitag, Gillian I Rice, Ashish Dhir, Marie-Louise Frémond, Mathieu P Rodero, Luis Seabra, Edwin Carter, Christine Bodemer, Daniela Buhas, Bert Callewaert, Pascale de Lonlay et al.
J Exp Med, 2021 Oct 4, PMID: 34387651 PMCID: PMC8374862 DOI: 10.1084/jem.20201560
2020
– Palliative Care in SMA Type 1: A Prospective Multicenter French Study Based on Parents » Reports.
Marie Hully, Christine Barnerias, Delphine Chabalier, Sophie Le Guen, Virginie Germa, Elodie Deladriere, Catherine Vanhulle, Jean-Marie Cuisset, Brigitte Chabrol, Claude Cances, Carole Vuillerot, Caroline Espil, Michele Mayer, Marie-Christine Nougues, Pascal Sabouraud, Jeremie Lefranc, Vincent Laugel, Francois Rivier, Ulrike Walther Louvier, Julien Durigneux, Sylvia Napuri, Catherine Sarret, Michel Renouil, Alice Masurel, Marcel-Louis Viallard, Isabelle Desguerre
Front Pediatr, 2020 Feb 18, PMID: 32133329 PMCID: PMC7039815 DOI: 10.3389/fped.2020.00004
– JAK Inhibition in the Aicardi-Goutières Syndrome.
Bénédicte Neven, Buthaina Al Adba, Marie Hully, Isabelle Desguerre, Claire Pressiat, Natalie Boddaert, Darragh Duffy, Gillian I Rice, Luis Seabra, Marie-Louise Frémond, Stéphane Blanche, Yanick J Crow
N Engl J Med, 2020 Nov 26, PMID: 33252884 DOI: 10.1056/NEJMc2031081
– Expanding the phenotype of mitochondrial disease: Novel pathogenic variant in ISCA1 leading to instability of the iron-sulfur cluster in the protein.
E Lebigot, M Hully, L Amazit, P Gaignard, T Michel, M Rio, M Lombès, P Thérond, A Boutron, M P Golinelli-Cohen
Mitochondrion, 2020 May, PMID: 32092383 DOI: 10.1016/j.mito.2020.02.008
2017
– Ciliogenesis and cell cycle alterations contribute to KIF2A-related malformations of cortical development.
Broix L, Asselin L, Silva CG, Ivanova EL, Tilly P, Gilet JG, Lebrun N, Jagline H, Muraca G, Saillour Y, Drouot N, Reilly ML, Francis F, Benmerah A, Bahi-Buisson N, Belvindrah R, Nguyen L, Godin JD, Chelly J, Hinckelmann MV.
Hum Mol Genet. 2017 Oct 25.
– Lower incidence of fracture after IV bisphosphonates in girls with Rett syndrome and severe bone fragility.
Lambert AS, Rothenbuhler A, Charles P, Brailly-Tabard S, Trabado S, Célestin E, Durand E, Fontaine I, Miladi L, Wicart P, Bahi-Buisson N, Linglart A.
PLoS One. 2017 Oct 26;12(10):e0186941.
– WDR81 mutations cause extreme microcephaly and impair mitotic progression in human fibroblasts and Drosophila neural stem cells.
Cavallin M, Rujano MA, Bednarek N, Medina-Cano D, Bernabe Gelot A, Drunat S, Maillard C, Garfa-Traore M, Bole C, Nitschké P, Beneteau C, Besnard T, Cogné B, Eveillard M, Kuster A, Poirier K, Verloes A, Martinovic J, Bidat L, Rio M, Lyonnet S, Reilly ML, Boddaert N, Jenneson-Liver M, Motte J, Doco-Fenzy M, Chelly J, Attie-Bitach T, Simons M, Cantagrel V, Passemard S, Baffet A, Thomas S, Bahi-Buisson
Brain. 2017 Oct 1;140(10):2597-2609.
– Genetics and mechanisms leading to human cortical malformations.
Romero DM, Bahi-Buisson N, Francis F.
Semin Cell Dev Biol. 2017 Oct 10.
– Cryo-EM Reveals How Human Cytoplasmic Dynein Is Auto-inhibited and Activated.
Zhang K, Foster HE, Rondelet A, Lacey SE, Bahi-Buisson N, Bird AW, Carter AP.
Cell. 2017 Jun 15;169(7):1303-1314.e18. doi: 10.1016/j.cell.2017.05.025. Epub 2017 Jun 8.
– Neuropathological Hallmarks of Brain Malformations in Extreme Phenotypes Related to DYNC1H1 Mutations.
Laquerriere A, Maillard C, Cavallin M, Chapon F, Marguet F, Molin A, Sigaudy S, Blouet M, Benoist G, Fernandez C, Poirier K, Chelly J, Thomas S, Bahi-Buisson
J Neuropathol Exp Neurol. 2017 Mar 1;76(3):195-205.
2016
– Mutations in the HECT domain of NEDD4L lead to AKT-mTOR pathway deregulation and cause periventricular nodular heterotopia.
Broix L, Jagline H, L Ivanova E, Schmucker S, Drouot N, Clayton-Smith J, Pagnamenta AT, Metcalfe KA, Isidor B, Louvier UW, Poduri A, Taylor JC, Tilly P, Poirier K, Saillour Y, Lebrun N, Stemmelen T, Rudolf G, Muraca G, Saintpierre B, Elmorjani A; Deciphering Developmental Disorders study., Moïse M, Weirauch NB, Guerrini R, Boland A, Olaso R, Masson C, Tripathy R, Keays D, Beldjord C, Nguyen L, Godin J, Kini U, Nischké P, Deleuze JF, Bahi-Buisson N, Sumara I, Hinckelmann MV, Chelly J.
Nat Genet. 2016 Nov;48(11):1349-1358. doi: 10.1038/ng.3676.
– Tubulinopathies Overview.
Bahi-Buisson N, Cavallin M. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K, editors.
GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2017.2016 Mar 24.
2014
– Mutations in tubulin genes are frequent causes of various foetal malformations of cortical development including microlissencephaly.
Fallet-Bianco C, Laquerrière A, Poirier K, Razavi F, Guimiot F, Dias P, Loeuillet L, Lascelles K, Beldjord C, Carion N, Toussaint A, Revencu N, Addor MC, Lhermitte B, Gonzales M, Martinovich J, Bessieres B, Marcy-Bonnière M, Jossic F, Marcorelles P, Loget P, Chelly J, Bahi-Buisson
Acta Neuropathol Commun. 2014 Jul 25;2:69.
– The wide spectrum of tubulinopathies: what are the key features for the diagnosis?
Bahi-Buisson N, Poirier K, Fourniol F, Saillour Y, Valence S, Lebrun N, Hully M, Bianco CF, Boddaert N, Elie C, Lascelles K, Souville I; LIS-Tubulinopathies Consortium, Beldjord C,
Chelly J.2014 Jun;137(Pt 6):1676-700.
– Mutations in Eml1 lead to ectopic progenitors and neuronal heterotopia in mouse and human.
Kielar M, Tuy FP, Bizzotto S, Lebrand C, de Juan Romero C, Poirier K, Oegema R, Mancini GM, Bahi-Buisson N, Olaso R, Le Moing AG, Boutourlinsky K, Boucher D, Carpentier W, Berquin P, Deleuze JF, Belvindrah R, Borrell V, Welker E, Chelly J, Croquelois A, Francis F
Nat Neurosci. 2014 Jul;17(7):923-33.
Cinq services hospitaliers de neuropédiatrie – dont celui de l’hôpital Necker avec les Pr Isabelle Desguerre, Pr Nadia Bahi-Buisson et Dr Marie Hully – et neuf établissements médico-sociaux qui accueillent des enfants atteints de polyhandicaps ont participé à une expérience pilote de télémédecine.
Ce film présente les bénéfices forts de cette expérience qui a apporté un renforcement du lien et du réseau de professionnels, une meilleure interaction entre les équipes, pour un objectif unique : le bien-être du patient !
En espérant que cette expérimentation se poursuive et se déploie !
- Association française du syndrome d’Angelman (AFSA)
- Association française du syndrome de Rett (AFSR)
- Rett Syndrome Europe
- Association FOXG1 France
- CDKL5 alliance francophone
- Association Syndrome Aicardi (ASA)
- Association de défense des polyhandicapés (ADEPO)
- Association Tous ensemble pour agir (TEPA)
- Au Clair de Lune
- Handy Rare et Poly
- Groupe Polyhandicap France (GPF)
- Comité d’études, d’éducation et de soins auprès des personnes polyhandicapées (CESAP)
- Association Les Tout Petits (LTP)
- Fast France
- Fondation Saint-Jean de Dieu
- Association Notre Dame de Joye
- Envoludia
- Alliance Maladies Rares
Mon enfant ne sera pas comme les autres
Le Monde | 15.12.2016
> Lire la suite
Vous trouverez ci-dessous les consignes et recommandations concernant l’appui des établissements de santé et des professionnels de ville aux personnes en situation de handicap en établissement ou à domicile pour la prise en charge des patients COVID-19.
Elles s’adressent à tous les professionnels de santé qu’ils soient libéraux, salariés et en établissements de santé publics ou privés et sont coordonnées sur le territoire par chaque ARS qui met en place une cellule médico-sociale au niveau régional déclinée dans chaque département.
Une première fiche synthétise les moyens à mobiliser et à structurer par les professionnels de santé de ville et les établissements de santé afin d’assurer la continuité des soins et des prises en charge des personnes en situation de handicap. Il s’agit également de garantir la qualité et la sécurité de la prise en charge des patients COVID+ en situation de handicap, en soutien aux professionnels du secteur social et médico-social ainsi que de contribuer à la limitation de la propagation de l’épidémie.
Ce document est accompagné d’un ensemble de fiches-reflexes destinées aux centres 15 sur les spécificités de certaines situations de handicap.
Coordonnées du CRMR
Hôpital universitaire Necker-Enfants malades
> Service de neurologie pédiatrique
149 rue de Sèvres
75743 PARIS Cedex 15
> Livret d’accueil pédiatrique
![]()
À Necker, le centre de référence des déficiences intellectuelles et polyhandicaps de causes rares c’est …
* données pour l’année 2022