Centre de référence des maladies héréditaires du métabolisme de l'enfant et de l'adulte (MAMEA)
![]()
![]() Le centre de référence de maladies rares MAMEA prend en charge les enfants et les adultes atteints de maladies héréditaires du métabolisme.
Le centre de référence de maladies rares MAMEA prend en charge les enfants et les adultes atteints de maladies héréditaires du métabolisme.
Il développe depuis plusieurs années les traitements les plus récents, médicamenteux et diététiques, ainsi que des thérapies innovantes, pour les maladies par intoxication par les protéines et les sucres, les maladies énergétiques liées à un défaut d’utilisation du glycogène ou des acides gras. Mais aussi, les hypoglycémies liées aux maladies énergétiques, un défaut de la néoglucogénèse et les hyperinsulinismes, et d’autres maladies neurométaboliques (CDG, maladies peroxysomales, maladie de Lyes Nyhan, déficit en sérine…).
Le site de Necker travaille en étroite collaboration avec les centres de compétence, en particulier de Rouen et Caen, les autres centres de référence et les associations de patients sur l’ensemble du territoire national, la filière de santé G2M et les centres européens (metabERN).
Mots-clés : Aminoacidopathies (leucinose, phénylcétonurie, tyrosinémie, homocystinurie, alcaptonurie…), aciduries organiques (acidémie méthylmalonique, acidémie propionique, acidémie isovalérique), déficits du cycle de l’urée (OTC, CPS, NAGS, AAS, ASL, ARG…), anomalies du métabolisme du galactose et du fructose, déficits de l’oxydation des acides gras, déficits de la néoglucogenèse, déficits du métabolisme des corps cétoniques, glycogénoses, hypoglycémies induites par hyper-insulinisme, erreurs de la glycosylation des protéines, erreurs du métabolisme des polyols, les maladies des peroxysomes, maladies neurométaboliques (purines, serine, creatine…)
Pour prendre rendez-vous
Pédiatrie
Secrétariat du professeur de Lonlay
Tel. 01 44 49 48 52
> Envoyer un mail
Secrétariat du professeur Schiff et du docteur Pichard
Tel. 01 44 49 55 21
> Envoyer un mail
Secrétariat du docteur Brassier et du docteur Bouchereau
Tel. 01 44 49 47 31
> Envoyer un mail
Secrétariat du docteur Arnoux
Tel. 01 44 49 40 23
> Envoyer un mail
Adulte
Secrétariat du docteur Servais
Tel. 01 44 49 54 13
> Envoyer un mail
Secrétariat du docteur Dao
Tel. 01 44 49 54 58
> Envoyer un mail
En cas
d’urgence
Prenez connaissance des protocoles d’urgence par symptôme ou maladie : https://www.filiere-g2m.fr/urgences
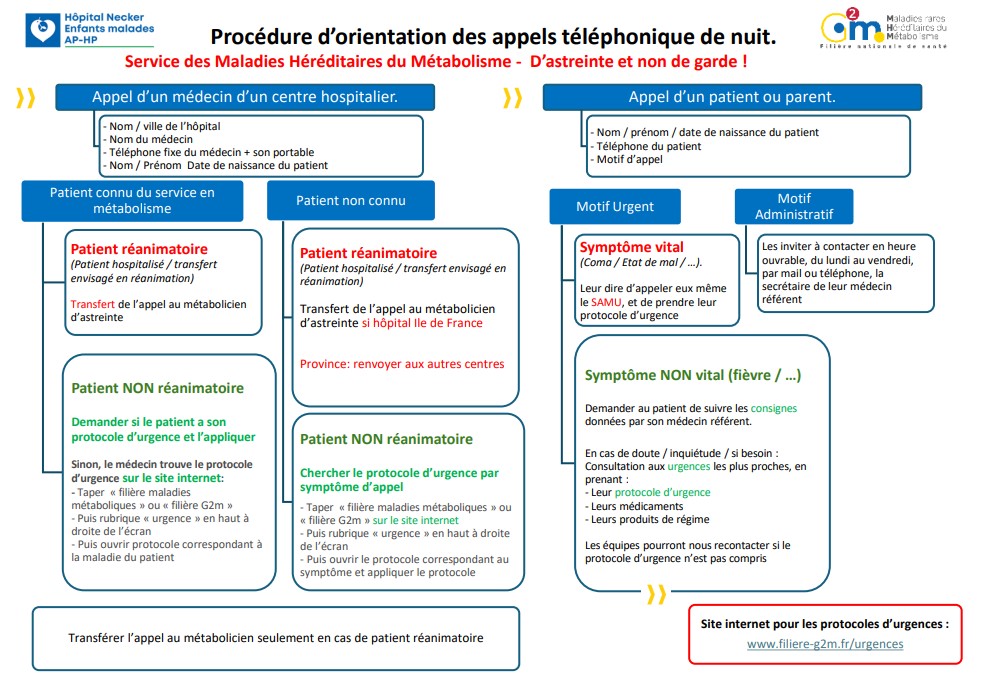
Procédure d’orientation des appels téléphonique de nuit :
- Pathologies et recommandations
- Voies métaboliques et pathologies
- Équipe médicale du CRMR
- Équipe paramédicale du CRMR
- Équipe administrative du CRMR
- Éducation thérapeutique
- Protocoles nationaux de diagnostic et de soins (PNDS)
- Recherche
- Enseignement
- Publications
- Associations de patients
- Carte du réseau national
- Evénements
- Revue de presse
Les maladies héréditaires du métabolisme (MHM) sont la conséquence du déficit d’origine génétique d’une enzyme ou d’un transporteur impliqués dans de nombreuses voies métaboliques.
Elles sont classées en 3 groupes selon une physiopathologie commune :
- Les maladies par intoxication
- Les maladies par déficit énergétique
- Les maladies des molécules complexes
Les MHM sont individuellement très rares (fréquence de 1/5 000 à 1/500 000) mais restent néanmoins très nombreuses puisqu’il est admis que sur les 4 000 à 6 000 maladies potentiellement existantes, seuls 500 environ sont actuellement identifiées avec de nouvelles descriptions de maladies parfois difficiles à classer.
Les deux premiers groupes sont aussi souvent désignés comme des anomalies du métabolisme intermédiaire, impliquant le métabolisme des protéines, des sucres, des lipides et ce sont ces maladies qui sont à risque de décompensation aigue.
Ces « détresses métaboliques » peuvent se présenter à tout âge, sous différentes formes.
Il est très important de les évoquer car la plupart sont traitables, le traitement sauve la vie à court terme et change le pronostic, notamment neurologique, à moyen et plus long terme.
Au delà de la démarche diagnostique et thérapeutique qui sont souvent mêlées et complexes au vu de la rareté des pathologies, il est primordial d’y penser et de savoir mettre en oeuvre les premières mesures simples avant de confier le patient à des équipes expérimentées, au sein des centres de référence et de compétence.
Les MHM représentent un défi non seulement pour le patient par la prise en charge, notamment en cas d’urgence métabolique, mais aussi pour la famille par le conseil génétique, le diagnostic prénatal à l’échelle individuelle.
A l’échelle collective la question d’étendre le dépistage néonatal de certaines MHM se pose, notamment grâce aux possibilités techniques actuelles, à la meilleure connaissance de l’histoire naturelle des maladies et au développement de thérapeutiques spécifiques, cette réflexion étant guidée par l’éthique.
Les maladies par intoxication
Ce groupe inclut les maladies du métabolisme intermédiaire qui entraînent une intoxication aigue ou progressive par l’accumulation de composés toxiques en amont du bloc enzymatique.
Ce groupe comprend les aminoacidopathies (phénylcétonurie, leucinose, homocystinurie, tyrosinémie…), les aciduries organiques (acidurie mathylmalonique, propionique, isovalérique…), les déficits du cycle de l’urée et apparentés (déficit en OTC, intolérance aux protéines dibasique…), les intolérances au sucre (galactosémie, intolérance héréditaire au fructose…).
Toutes ces affections, à quelques exceptions près, partagent des caractéristiques communes.
Elles n’interfèrent pas avec le développement embryo-fœtal et se présentent après un intervalle libre après la naissance par des signes cliniques d’intoxication aigue (vomissement, léthargie, coma, défaillance multiviscérale…) ou chronique (anorexie, retard de croissance, retard psychomoteur, cardiomyopathie…).
Elles sont susceptibles de décompenser en « crises métaboliques », de façon récurrente à l’occasion d’évènements cataboliques (fièvre, infection intercurrente, jeûne…) ou lors de l’ingestion d’aliments « toxiques ».
Leur diagnostic repose sur les chromatographies des acides aminés sanguins et urinaires, organiques urinaires et le profil des acylcarnitines. La plupart de ces maladies sont traitables par l’épuration des composés toxiques en situation d’urgence (hémodialyse, médicaments épurateurs…) et par des régimes spéciaux restrictifs le reste du temps, à vie.
Les maladies par déficit énergétique
Ce groupe rassemble les maladies héréditaires du métabolisme avec des symptômes liés, au moins en partie, au défaut de production, utilisation ou stockage de l’énergie, impliquant le foie, le muscle strié (périphérique et cardiaque), le cerveau, la rétine…forts consommateurs d’énergie.
Il peut être schématiquement divisé en 2 en distinguant le défaut du métabolisme énergétique mitochondrial et cytoplasmique.
Les déficits énergétiques mitochondriaux sont les plus sévères et généralement non traitables.
Ils comprennent les acidoses lactiques congénitales (déficit du transporteur mitochondrial du pyruvate, déficit en pyruvate carboxylase, déficit en pyruvate déshydrogénase et déficits du cycle de Krebs), les déficits de la chaîne respiratoire (impliquant les 5 complexes de la chaîne respiratoire mais aussi les transporteurs mitochondriaux des molécules énergétiques et la synthèse du coenzyme Q10), les déficits de l’oxydation des acides gras et le métabolisme des corps cétoniques.
Les déficits énergétiques cytoplasmiques sont généralement moins graves.
Ils comprennent les déficits du métabolisme de la glycolyse (voie des pentoses..) et du glycogène (néoglucogénèse et glycogénolyse avec les glycogénoses hépatiques et musculaires), les hyperinsulinismes, les désordres du métabolisme de la créatine.
Les signes d’appel communs sont hypoglycémie, hépatomégalie, acidose lactique, myo(cardio)pathie, retard de croissance, hypotonie ou symptômes neurologiques variés, mort subite dans l’enfance.
Certains déficits mitochondriaux et déficits sur la voie des pentoses peuvent interférer avec le développement embryo-fœtal et entraînent une dysmorphie, des dysplasies et malformations.
Le diagnostic est difficile et repose sur des explorations fonctionnelles (épreuves de jeûne…), dosages enzymatiques nécessitant des cultures cellulaires (biopsies de peau, muscle…) et sur des analyses moléculaires.
Les maladies des molécules complexes
Ce groupe concerne les organelles intracellulaires (lysosome, peroxysome…) et regroupe les maladies qui perturbent la synthèse ou le catabolisme des molécules complexes au sein de celles-ci : les maladies lysosomales, peroxysomales, les déficits de la glycosylation des protéines (CDG syndromes), les déficits de la synthèse endogène du cholestérol et des acides biliaires ainsi-que d’autres déficits impliquant le trafic des molécules complexes.
Les symptômes sont permanents, progressifs et indépendants des évènements intercurrents ou de l’alimentation puisqu’ils concernent les protéines de structure et non celles impliquées dans les voies du métabolisme intermédiaire.
Ces maladies sont polymorphes, multiorganiques, souvent à expression neurologique, pouvant entraîner des atteintes dès la vie fœtale (anasarques foeto-placentaire, malformations cérébrales, syndromes polymalformatifs…);
Le diagnostic repose sur des dosages enzymatiques spécifiques pouvant être basés sur des test urinaires de dépistage et la mise en évidence de mutations en biologie moléculaire.
La plupart de ces maladies ne sont pas traitables mais il existe à l’heure actuelle l’enzymothérapie substitutive pour certaines d’entre elles et un large champs de recherche consacré à l’essor de nouvelles thérapeutiques.

![]()
Métabolisme énergétique
Maladies mitochondriales
- Anomalie de la phosphorylation oxydative mitochondriale due à des anomalies de l’ADN nucléaire
- Déficit en coenzyme Q10
- Anomalie de la phosphorylation oxydative mitochondriale due à des anomalies de l’ADN mitochondrial
- Syndrome de Kearns-Sayre
- Syndrome de Pearson
- MELAS
- MERRF
- Neuropathie optique héréditaire de Leber
- Syndrome NARP
- Diabète-surdité de transmission maternelle
- Anomalie isolée d’un complexe de la phosphorylation oxydative
- Anomalie de la phosphorylation oxydative mitochondriale « non caractérisée »
Défaut de biosynthèse de l’acide lipoïque
Déficit en Créatine
- Déficit en guanidinoacétate méthyltransférase
- Déficit en l-arginine: glycine amidinotransférase
- Déficit en transporteur de la créatine lié à l’X
Trouble de l’oxydation des acides gras et du métabolisme des corps cétoniques
- Trouble de la cétolyse
- Déficit en bêta-cétothiolase
- Déficit en succinyl-CoA:3-cétoacide CoA transférase
- Trouble de l’oxydation des acides gras et de la cétogénèse
- Déficit en protéine trifonctionnelle mitochondriale
- Déficit en 3-hydroxy-3-méthylglutaryl-CoA-Lyase
- Déficit en 3-hydroxy-3-méthylglutaryl-CoA synthétase
- Déficit en acyl-CoA déshydrogénase des acides gras à chaîne moyenne
- Déficit multiple en acyl-CoA déshydrogénases
- Déficit en acyl-CoA déshydrogénase des acides gras à chaîne courte
- Déficit en acyl-CoA déshydrogénase des acides gras à chaîne très longue
- Déficit en 3-hydroxyacyl-CoA déshydrogénase des acides gras à chaîne longue
- Déficit en 3-hydroxylacyl-CoA déshydrogénase des acides gras à chaîne courte
- Déficit en carnitine palmitoyltransférase II
- Déficit en carnitine palmitoyltransférase 1A
- Déficit systémique primaire en carnitine
- Déficit en carnitine-acylcarnitine translocase
- Acidurie malonique
- Déficit en acyl-CoA déshydrogénase 9
- Déficit en transporteur 1 de monocarboxylate Anomalie du métabolisme du pyruvate
- Déficit en pyruvate déshydrogénase
- Déficit en pyruvate kinase du globule rouge
- Anomalie du Cycle de Krebs
- Acidurie oxoglutarique
Métabolisme des acides aminés
Trouble de l’absorption et du transport des acides aminés
- Cystinurie
- Hyperaminoacidurie dicarboxylique
- Intolérance aux protéines dibasiques avec lysinurie
- Aminoacidurie hyperdibasique type 1
- Syndrome d’hypotonie-cystinurie
- Maladie de Hartnup
- Iminoglycinurie
Trouble du métabolisme du cycle de l’urée et de la détoxification de l’ammoniac
- Déficit en ornithine transcarbamylase
- Argininémie
- Acidurie argininosuccinique
- Déficit en carbamoyl-phosphate synthétase 1
- Citrullinémie type I
- Citrullinémie type II
- Cholestase intrahépatique néonatale par déficit en citrine
- Syndrome d’hyperornithinémie-hyperammoniémie-homocitrullinurie
- Déficit en N-acétylglutamate synthase
- Syndrome d’hyperinsulinisme et hyperammoniémie
- Déficit en anhydrase carbonique VA
Trouble du métabolisme du cycle de la méthionine, des acides aminés sulfurés et cobalamine
- Homocystinurie classique par déficit en cystathionine bêta-synthase / Lien Orphanet
- Déficit en sulfite oxydase
- Homocystinurie sans acidurie méthylmalonique
- Homocystinurie par déficit en méthylène tétrahydrofolate réductase + MTHF
- Déficit en méthylcobalamine type cblE
- Déficit en méthylcobalamine type cblG
- Déficit en méthylcobalamine type cblDv1
- Hyperméthioninémie par déficit en adénosine kinase
- Hyperméthioninémie par déficit en glycine N-méthyltransférase
- Acidémie méthylmalonique avec homocystinurie type cblC / Lien Orphanet
- Acidémie méthylmalonique avec homocystinurie type cblD / Lien Orphanet
- Acidémie méthylmalonique avec homocystinurie type cblF / Lien Orphanet
- Acidémie méthylmalonique avec homocystinurie type cblJ / Lien Orphanet
- Acidémie méthylmalonique avec homocystinurie type cblX / Lien Orphanet
- Acidémie méthylmalonique sensible à la vitamine B12 type cblA
- Acidémie méthylmalonique sensible à la vitamine B12 type cblB
- Acidémie méthylmalonique sensible à la vitamine B12 type cblDv2
- Acidurie formiminoglutamique
- Syndrome neurodégénératif dû au déficit de transport cérébral des folates
- Maladie des noyaux gris centraux sensible à la biotine par déficit en transporteur de la thiamine
- Encéphalopathie par déficit en thiamine pyrophosphokinase
Trouble du métabolisme de l’histidine
Trouble du métabolisme de la proline
Trouble du métabolisme de l’ornithine / Lien orphanet
- Atrophie gyrée choriorétinienne
Trouble du métabolisme des peptides
- Carnosinémie
- Déficit en prolidase
- Homocarnosinose
Trouble du métabolisme de phenylalanine / Lien orphanet
- Phénylcétonurie classique
- Hyperphénylalaninémie modérée permanente
- Phénylcétonurie atypique
- Embryo-foetopathie liée à une PCU maternelle
Trouble du métabolisme de la tyrosine
- Alcaptonurie
- Hawkinsinurie
- Tyrosinémie type 1
- Tyrosinémie type 2
- Tyronsinémie type 3
Trouble du métabolisme de la sérine ou de la glycine
- Sacrosinémie
- Hyperglycinémie sans cétose
- Maladie neurométabolique par déficit en sérine
- Déficit en 3-phosphoglycerate déshydrogénase
- Déficit en 3-phosphoglycerate deshydrogénase, forme infantile/juvénile
Trouble du cycle du gamma-glutamyl
- Déficit en glutathion synthétase
- Déficit en 5-oxopolinase
- Déficit en gamma-glutamyl transpeptidase
- Déficit en gamma-glutamylcystéine synthétase
Trouble du métabolisme des acides aminés à chaînes ramifiées
- Leucinose / Lien Orphanet
- Déficit en méthylmalonate semialdéhyde déshydrogénase
- Déficit en kinase déshydrogénase des cétoacides à chaînes ramifiées
Trouble du métabolisme du tryptophane
- Hydroxykynuréninurie
Trouble du métabolisme de la lysine et de l’hydroxylysine
- Hyperlysinémie
- Saccharopinurie
- Acidurie 2-aminoadipique 2-oxoadipique
- Hydroxylysinurie
Trouble du métabolisme de la glutamine
Acidurie organique
- Acidurie L-2-hydroxyglutarique
- Acidurie D-2-hydroxyglutarique
- Acidurie D,L-2-hydroxyglutarique
- Déficit en glutaryl-CoA déshydrogénase
- Maladie de Canavan
- Déficit en aminoacylase 1
- Maladie HSD10
- Acidémie isovalérique
- Déficit en biotinidase / Lien Orphanet
- Déficit en holocarboxylase synthétase
- Déficit en bêta-cétothiolase
- Acidurie 3-hydroxyisobutyrique
- Acidurie 3-hydroxy-3-méthylglutarique
- Déficit en 3-méthylcrotonyl-CoA carboxylase
- Acidémie propionique / Lien Orphanet
- Déficit en 2-méthylbutyryl-CoA déshydrogénase
- Déficit en isobutyryl-CoA déshydrogénase
- Déficit en 3-hydroxyisobutyryl-CoA hydrolase
- Acidémie combinée malonique et méthylmalonique
- Acidurie 3-methylglutaconique
- Syndrome de Barth
- Acidurie 3-méthylglutaconique type 1
- Acidurie 3-méthylglutaconique type 3
- Acidurie 3-méthylglutaconique type 4
- Syndrome MEGDEL
- Acidurie 3-méthylglutaconique type 7
Acidémie méthylmalonique sans homocystinurie / Lien Orphanet
- Acidémie méthylmalonique résistante à la vitamine B12
- Acidémie méthylmalonique isolée résistante à la vitamine B12 type mut-
- Acidémie méthylmalonique résistante à la vitamine B12 type mut0
- Acidémie méthylmalonique sensible à la vitamine B12
- Acidémie méthylmalonique par déficit en méthylmalonyl-CoA épimérase
- Déficit en transaminase de l’acide gamma-aminobutyrique
Métabolisme des glucides
- Trouble du métabolisme des hydrates de carbone
- Déficit en fructose-1,6 diphosphatase
- Déficit en phosphoénolpyruvate carboxykinase
- Déficit en pyruvate carboxylase
- Déficit isolé en glycérol kinase
- Glycogénose par déficit en enzyme débranchante (glycogénose de type III)
- Glycogénose par déficit en enzyme branchante
- Glycogénose par déficit en glycogène phosphorylase musculaire
- Glycogénose par déficit en phosphorylase hépatique
- Glycogénose par déficit en phosphofructokinase musculaire
- Glycogénose par déficit en glucose-6-phosphatase (glycogénose de type I) / Lien Orphanet
- Glycogénose par déficit en glucose-6-phosphatase de type a
- Glycogénose par déficit en glucose-6-phosphatase de type b
- Glycogénose par déficit en aldolase A musculaire
- Glycogénose par déficit en phosphoglycérate kinase 1
- Glycogénose par déficit en GLUT2
- Glycogénose par déficit en lactate déshydrogénase
- Glycogénose par déficit en phosphoglycérate mutase
- Glycogénose par déficit en bêta-énolase musculaire
- Glycogénose par déficit en glycogénine
- Glycogénose par déficit en glycogène synthase hépatique (glycogénose de type 0)
- Glycogénose par déficit en glycogène synthase cardiaque et musculaire
- Déficit en triose-phosphate isomérase
- Déficit en phosphoglucose isomérase
- Hyperinsulinisme par déficit en glucokinase
- Intolérance au fructose héréditaire
- Déficit en galactokinase
- Déficit en galactose épimérase
- Galactosémie classique
- Acidurie D-glycérique
- Hyperoxalurie primitive
- Déficit congénital en saccharase-isomaltase
- Déficit congénital en lactase
- Malabsorption du glucose-galactose
- Déficit en GLUT1
- Déficit en transaldolase
Métabolisme lysosomal
Céroïde-lipofuscinose neuronale
- Céroïde-lipofuscinose neuronale de l’adulte
- Céroïde-lipofuscinose neuronale infantile
- Céroïde-lipofuscinose neuronale juvénile
- Céroïde-lipofuscinose neuronale congénitale
- Céroïde-lipofuscinose neuronale infantile tardive
- Déficit en phosphatase acide
Anomalie de transport lysosomal des acides aminés
- Cystinose
- Maladie de surcharge en acide sialique libre
Mucopolysaccharidose
- Mucopolysaccharidose type 6
- Mucopolysaccharidose type 7
- Mucopolysaccharidose type 2
- Mucopolysaccharidose type 1 / Lien Orphanet
- Mucopolysaccharidose type 3
- 3A
- 3B
- 3C
- 3D
- Mucopolysaccharidose type 4
- 4A
- 4B
- Déficit en hyaluronidase
Sphingolipidose
- Déficit multiple en sulfatases
- Maladie de Farber
- Maladie de Krabbe
- Maladie de Fabry / Lien Orphanet
- Leucodystrophie métachromatique
- Maladie de Gaucher / Lien Orphanet
- Maladie de Niemann-Pick type A
- Maladie de Niemann-Pick type B
- Maladie de Niemann-Pick type C / Lien Orphanet
- Déficit en lipase acide lysosomale
- Maladie de Niemann-Pick type E
- Déficit en prosaposine
- Gangliosidose à GM1
- Gangliosidose à GM2
- Mucolipidose type II
- Mucolipidose type III
- Alpha-mannosidose
- Aspartylglucosaminurie
- Bêta-mannosidose
- Fucosidose
- Galactosialidose
- Déficit en alpha-N-acétylgalactosaminidase
- Sialidose type 1
- Sialidose type 2
- Sialurie
- Maladie de Wolman
- Maladie de stockage des esters du cholestérol
Maladie de surcharge lysosomale du glycogène
- Glycogénose par déficit en maltase acide
- Glycogénose par déficit en LAMP-2
Métabolisme péroxysomal
- Adrénoleucodystrophie néonatale
- Maladie de Refsum infantile
- Maladie de Refsum / Lien Orphanet
- Acatalasémie
- Acidémie glutarique type 3
- Déficit congénital de synthèse des acides biliaires type 4
- Déficit en acyl-CoA oxydase
- Adrénoleucodystrophie liée à l’X / Lien Orphanet
- Déficit en enzyme bifonctionnelle
- Hyperoxalurie primitive type 1
- Chondrodysplasie ponctuée rhizomélique
Métabolisme des stérols et des lipoprotéines
Défaut de synthèse des acides biliaires avec cholestase et malabsorption
- Xanthomatose cérébrotendineuse / Lien Orphanet
- Déficit congénital de synthèse des acides biliaires type 4
- Hypercholestérolémie due au déficit en cholesterol 7alpha-hydroxylase
- Lathostérolose
- Syndrome de Smith-Lemli-Opitz / Lien Orphanet
- Desmostérolose
- Acidurie mévalonique / Lien Orphanet
Dyslipidémie rare
- Hyperlipidémie rare
- Hyperlipidémie combinée
- Hyperlipoprotéinémie type 3
- Hypertriglycéridémie majeure
- Hyperalphalipoprotéinémie
- Déficit en LCAT
- Déficit en apolipoprotéine A-I
- Maladie de Tangier
Hypobêtalipoprotéinémie
- Abêtalipoprotéinémie
- Maladie de rétention des chylomicrons
- Sitostérolémie
Autres lipidoses
- Syndrome de Sjögren-Larsson
- Maladie de Dorfman-Chanarin
Glycosylation des protéines
- Anomalie de la N-glycosylation (CDG Ia – CDG X) / Anomalie de la N-glycosylation sur Orphanet
- Anomalie de la O-glycosylation
- Anomalie de synthèse des O-xylosylglycanes
- Maladie des exostoses multiples
- Syndrome d’Ehlers-Danlos type musculo-contractural
- Syndrome d’Ehlers-Danlos type progéroïde
- Anomalies multiples de la glycosylation
- Anomalie du complexe oligomérique du Golgi
Métabolisme des purines et pyrimidines
Déficit en hypoxanthine-guanine phosphoryl transférase
- Syndrome de Lesch-Nyhan / Syndrome de Lesch-Nyhan sur Orphanet
- Déficit partiel en hypoxanthine guanine phosphoribosyltransférase
Trouble du métabolisme de la pyrimidine
- Acidurie orotique héréditaire
- Déficit en dihydropyrimidine déshydrogénase
- Dihydropyrimidinurie
- Déficit en bêta-uréidopropionase
Neurotransmission
Anomalie des neurotransmetteurs
- Déficit en monoamine oxydase A
- Déficit en dihydroptéridine réductase / Déficit en dihydroptéridine réductase sur Orphanet
- Déficit en 6-pyruvoyl-tétrahydroptérine synthase
- Déficit en GTP cyclohydrolase I
- Déficit en dopamine bêta-hydroxylase
- Déficit en décarboxylase des acides aminés aromatiques
- Déficit en tyrosine hydroxylase
- Déficit du transport de la dopamine
- Acidurie 4-hydroxybutyrique
- Déficit en transaminase de l’acide gamma-aminobutyrique
- Epilepsie B6 et B9-dépendante par déficit en antiquitin
- Convulsions sensibles au phosphate de pyridoxal
- Déficit en adénylosuccinate lyase
- Déficit en adénosine phosphoribosyltransférase
Dr Jean-Baptiste Arnoux
Dr Anaïs Brassier
Dr Juliette Bouchereau
Dr Samia Pichard
Dr Claire Marine Berat
Médecins adultes
Dr Aude Servais
Dr Myriam Dao
Biologistes
Pr Jean François Benoist
Pr Chris Ottolenghi
Pr Robert Barouki
Dr Odile Rigal
Dr Clément Pontoizeau
Dr Apolline Imbard
Dr Allel Chabli
Dr Catherine Caillaud
Dr Sylvia Sanquer
Dr Edouard Le Guillou
Diététiciens (nes)
Murielle Assoun
Claire Belloche
Sandrine Dubois
Florence Serceau
Camille Baini
Soraya Shafieivand
Sabine Dewulf
Bénédicte Samba
Psychologue
Valérie Barbier
Neuropsychologue
Fanny Gadet
Ergothérapeutes
Élodie Deladrière
Enseignante
Anne-Cécile Causse
Psychomotricienne
Manon Tessier
Assistante sociale
Virginie Leboeuf
Educatrice jeunes enfants
Manuella Bayart
Masseuse kinésithérapeute
Marion Maquet
Secrétaires
Luisa Paciencia
Ketty Dino
Nathalie Madouri
Sophie Bustos
Victoire Ewande
Nathalie Mandane
Isabelle Vincent
Isabelle Madelaine
Yannick Asdrubal
- Intitulé du programme : Programme d’éducation thérapeutique « Ma santé pas à pas »
Contact : Dr Manuel SCHIFF (Envoyer un mail)
Localisation : Centre de référence des maladies métaboliques MaMEA, Hôpital Necker Enfants malades, Paris
Informations complémentaires : Enfants à partir de 5 ans et adultes
- Intitulé du programme : Programme d’éducation thérapeutique pour les maladies héréditaires du métabolisme : Déficits du cycle de l’urée, aciduries organiques, leucinose et acidurie glutarique de type I
Contact : Dr Pascale de LONLAY (Envoyer un mail)
Localisation : Centre de référence des maladies métaboliques MaMEA, Hôpital Necker Enfants malades, Paris
Informations complémentaires : Enfants de 0 à 18 ans et leurs parents
- Intitulé du programme : Programme d’éducation thérapeutique pour les maladies héréditaires du métabolisme : Déficit en PDH, Glut-1 et cytopathie mitochondriale
Contact : Dr Pascale de LONLAY (Envoyer un mail)
Localisation : Centre de référence des maladies métaboliques MaMEA, Hôpital Necker Enfants malades, Paris
Informations complémentaires : Enfants jusque 18 ans et leurs parents
- Intitulé du programme : Programme d’éducation thérapeutique pour les maladies héréditaires du métabolisme : Déficit de l’oxydation des acides gras
Contact : Dr Pascale de LONLAY (Envoyer un mail)
Localisation : Centre de référence des maladies métaboliques MaMEA, Hôpital Necker Enfants malades, Paris
Informations complémentaires : Enfants jusque 18 ans et leurs parents
- Intitulé du programme : Programme d’éducation thérapeutique pour les maladies héréditaires du métabolisme : Galactosémie et fructosémie
Contact : Dr Pascale de LONLAY (Envoyer un mail)
Localisation : Centre de référence des maladies métaboliques MaMEA, Hôpital Necker Enfants malades, Paris
Informations complémentaires : Enfants de 0 à 18 ans et leurs parents
- Intitulé du programme : Programme d’éducation thérapeutique pour les maladies héréditaires du métabolisme : Glycogénoses et déficits de la néoglucogenèse
Contact : Dr Pascale de LONLAY (Envoyer un mail)
Localisation : Centre de référence des maladies métaboliques MaMEA, Hôpital Necker Enfants malades, Paris
Informations complémentaires : Enfants jusque 18 ans et leurs parents
- Intitulé du programme : Programme d’éducation thérapeutique pour les maladies héréditaires du métabolisme à traitement diététique : Hyper-Insulinisme
Contact : Dr Pascale de LONLAY (Envoyer un mail)
Localisation : Centre de référence des maladies métaboliques MaMEA, Hôpital Necker Enfants malades, Paris
Informations complémentaires : Enfants jusque 18 ans et leurs parents
- Intitulé du programme : Programme d’éducation thérapeutique pour les maladies héréditaires du métabolisme : Phénylcétonurie, Tyrosinémie, Homocystinurie
Contact : Dr Pascale de LONLAY (Envoyer un mail)
Localisation : Centre de référence des maladies métaboliques MaMEA, Hôpital Necker Enfants malades, Paris
Informations complémentaires : Enfants de 0 à 18 ans et leurs parents
- PNDS Mucopolysaccharidoses (actualisation en cours)
- PNDS Maladie de Pompe (actualisation en cours)
- PNDS Alpha-mannosidose (en cours)
- PNDS Acidémie isovalérique (en cours)
- PNDS Anomalies du métabolisme du cuivre (hors maladie de Wilson) (en cours)
- PNDS Encéphalopathie myo-neuro-gastrointestinale (en cours)
- PNDS Syndrome de Leigh (en cours)
- PNDS Maladie de Lesch-Nyhan (en cours)
- PNDS Déficit en sphingomyélinase acide Niemann PickA/B (en cours)
- PNDS Alcaptonurie en partenariat avec la filière OSCAR (en cours)
- PNDS Trouble de la remethylation (CBLC, METAB INTRACELL B12 ET MTHFR) (en cours)
- PNDS Maladie de Gaucher (2022) – révision par le CRMR MAMEA
- PNDS Diabète monogénique de type MODY (2022) – participation du CRMR MAMEA avec le CRMR PRISIS
- PNDS Tyrosinémie I (2022) – participation du CRMR MAMEA avec le CRMR PRISIS
- PNDS MPI-CDG Défaut de glycosylation des glycoprotéines par déficit en phosphomannose isomérase (2022) – coordonné par le CRMR MAMEA
- PNDS Homocystinurie par déficit en cytathionine-bêta-synthase (CBS) (2022) – coordonné par le CRMR MAMEA
- PNDS déficits de l’oxydation mitochondriale des acides gras (2021)
- PNDS Leucinose (2021)
- PNDS Hyperinsulinisme (2020)
- PNDS aciduries organiques (2020)
- PNDS maladie de McArdle et maladie glycogénose type V (2019) en collaboration avec FILNEMUS
- PNDS Phénylcétonurie (2018)
- PNDS MPS (2016)
- PNDS Maladie de Pompe (2016)
- PNDS Syndrome de Prader-Willi (2012)
- PNDS Maladie de Fabry (2010)
Recherche non-industriels en cours en 2022
Evaluation d’un traitement à l’allopurinol sur les troubles autistiques et l’épilepsie dans le déficit en adénylosuccinate lyase (ADSL) ; National, porteur du projet : P de Lonlay (N° EUDRACT 2017-002155-28) ; Financement : PHRC ADSL NCT 03776656, 2017, 170806 euros (jusque 2023)
MetabInf: role of mitophagy and innate immune system in the febrile decompensations of inherited rhabdomyolysis: therapeutic perspectives ; National, porteur du projet : P de Lonlay ; Financement : Agence Nationale de la Recherche (ANR Fr) : 2018-2024 (report) (500 000 euros)
TANGO2 deficiency: physiopathology and therapeutic perspective ; International, porteur du projet : P de Lonlay ; Financement : Fondation Tango2 (USA) 2020, 50 000 euros
Acute Rhabdomyolysis and Muscle Pain Associated With Mutations in the LPIN1 Gene – A Retrospective Study Describing the Safety and Efficacy of Hydroxychloroquine Sulfate Given on a Compassionate Basis to Patients Suffering From Lipin-1 Deficiency; National et international, porteur du projet : P de Lonlay ; Financement : Association Française pour la Myopathie (AFM) 2017, 36k€, Prix Necker 2017, 36k€, Prix Université Paris Centre 2017, 36k€, et industriel avec Imagine (2022 – ); NCT 04007562
Etude rétrospective de l’efficacité du solumedrol dans les rhabdomyolyses des patients avec un déficit en lipin-1 ; National, porteur du projet : P de Lonlay
AFM 2022 Autophagy dysregulation in acute rhabdomyolysis 2022, 200 k€
ANR – AAPG2023 projet Rhabdophagy : Déchiffrer et cibler les défauts d’autophagie dans la rhabdomyolyse, 533 223,00 €
Thérapie génique par AAV ciblant le foie pour la leucinose ; National, porteur du projet : M Schiff ; Financement : DIM Thérapie Génique Ile de France, 2018, 220 000 euros ; Brevet février 2020: EP20305079.4; thérapie génique de la leucinose dans un modèle murin, Inserm transfert 2020
Annonce des maladies rares du métabolisme dans le cadre du dépistage néonatal systématique : l’expérience de la phénylcétonurie ; National, porteur du projet : P de Lonlay et M Araneda (Université Paris) ; Financement : AAPSHS, Imagine et Maladies génétiques rares 2022, 33k€ et Filière salaire doctorant, 200 000 euros ; et Labex post-doc 2021 SHS
Personalized Mitochondrial Medicine (PerMiM): Optimizing diagnostics and treatment for patients with mitochondrial diseases – PerMiM ; Européen, porteur du projet : Professor Holger Prokisch, University of Munich ; Financement : ERA PerMed 2019 – APP transnational dans le cadre de l’ERA PerMed co-fund; projet ERARE, coord. Holger Prokisch (PerMIM https://anr.fr/Project-ANR-19-PERM-0006), 2020-2024, 250 000 euros
Evaluate the Effort Test as a Therapeutic Monitoring Tool in Acute Rhabdomyolyses ClinicalTrials.gov Identifier: NCT 03802279; Rhabdomyolysis Linked to a Hereditary Disease of Metabolism; National, porteur du projet : P de Lonlay
Etude des muccopolysaccharidoses RADICO (recherche transversale filière G2M)
National, porteur du projet : Dr Héron, maladies lysosomales, Trousseau ; Financement : RaDiCo-MPS n°MESR DC-2015-2482 –INSERM ; 80 patients pour Necker (A Brassier)
Etude prospective sur les conséquences du COVID-19 sur l’équilibre des MHM chez les patients contractant ou ayant contracté la COVID-19 : recherche transversale filière G2M; National, porteur du projet : Dr Claire Douillard, CRMR MHM Lille ; Financement : ARC filière, 2020 ; Patients ayant eu la Covid-19 ; Necker P de Lonlay : N° EUDRACT : 2020-A02886-33
Utilisation hors AMM du dapaglifozine (FORXIGA®) pour le traitement de la neutropénie dans les glycogénoses Ib : recherche transversale filière G2M et inter-filières ; National, porteur du projet : Jean Donadieu, CRMR neutropénie, Trousseau ; Financement : ARC filière, 2021
Maladies lysosomales C Caillaud (INEM) ; National, porteur du projet : C Caillaud ; Financement : INSERM ; Model murin
Obtention d’un PHRC Projet : AOM22231 – Pascale DE LONLAY, Titre du Projet : « Traitement des patients ayant un déficit en pyruvate déshydrogénase (PDH) par le phénylbutyrate de glycérol (RAVICTI®) », ; National multisites
Immunité dans la maladie de Pompe – Julien Diana et P de Lonlay (INEM)
(en cours écriture pour 2023 : demande PHRC PMM2-CDG et Diamox/Epalrestat)
En cours : Manganèse et galactose per os dans les CDG avec troubles de l’homéostasie golgienne
En cours : Impacts du monitoring continu de la glycémie chez les enfants atteints d’une glycogénose de type Ia et Ib et leurs parents (Acronyme : DEXCOM-PEDIATRIE)
Recherche industriels en cours en 2022
Interventionnels
• Mini comet Dimension territoriale du projet : international, Enzymothérapie Maladie de Pompe, Année du financement : 2019
• PEACE Dimension territoriale du projet : international, Enzymothérapie Pegzilarginase Effects on Arginase 1 deficiency Cinical Endpoints), Année du financement : 2020
• Tyre Sphere Dimension territoriale du projet : National, Tyrosinémie 1 – mélange d’acides aminés, Année du financement : 2021
• PTA Dimension territoriale du projet : International, Enzymothérapie Maladie de Pompe (suite Mini comet, phase 4), Année du financement : 2022 – en préparation 2021
• Cytopathies mitochondriales étude interventionnelle EPI-743, Dimension territoriale du projet : international, Traitement de l’épilepsie des cytopathies mitochondriales par quinones passant la BHM, Année du financement : 2021
• LEAP2MONO – EFC17215 : Etude de phase 3, multicentrique, internationale, randomisée, double blind, double-dummy, avec comparateur actif pour évaluer l’efficacité et la sécurité du venglustat chez les patients adultes et pédiatriques atteints de la maladie de Gaucher de type 3 (GD3) qui ont atteint les objectifs thérapeutiques avec l’Enzyme Thérapie de remplacement : nov 2022
Observationnels (registres)
• MPSI, Dimension territoriale du projet : international, Mucopolysaccharidosis I (MPS I) Registry
• Protect, Dimension territoriale du projet : international, Understanding the long-term management of Organic Acideamia Patients with CARBAGLU® Pharmerit HOS, Dimension territoriale du projet : international, A Global, Multi-Center, Long-Term, Observational Registry of Patients with Hunter Syndrome (Mucopolysaccharidosis Type II, MPS II)
• MPI-CDG (CDG-Ib) étude observationnelle mannose Cerecor, Dimension territoriale du projet : international, Etudedu mannose dans le CDG-Ib ou MPI-CD, Année du financement : 2019
• Déficits cycle urée étude observationnelle citrulline Biocodex, Dimension territoriale du projet : national, Etude à long-terme des déficits du cycle de l’urée traités par arginine et/ou citrulline, Année du financement : 2019
• Leucinose étude observationnelle acides aminés recordati, Dimension territoriale du projet : national, Etude du mélange d’acides aminés intra-veineux dans la leucinose, Année du financement : 2019
• PMM2-CDG étude observationnelle Glycomine, Dimension territoriale du projet : international, Etude de l’histoire naturelle des patients avec PMM2-CDG ou CDG Ia, Année du financement : 2019
• Ac sphingolipid deficiency (ASMD) étude observationnelle, Dimension territoriale du projet : international, Année du financement : 2020
• m-RNA -3704-P001 aciduries organiques étude observationnelle, Dimension territoriale du projet : international, Année du financement : 2019
• Hyperinsulinisme congénital : une étude longitudinale rétrospective (CHART) Dimension territoriale du projet : international, Année du financement : 2022
• Pompe Disease Registry
• Registre LAL
• Registre KAMPER, suivi du traitement par sapropterine dans la phénylcétonurie
• MPS VI étude observationnelle
• Gaucher Registry-International Collaborative Gaucher Group (ICGG)
Diplôme inter-universitaire maladies héréditaires du métabolisme
Université de Paris
2022
– Heterogeneity of PNPT1 neuroimaging: mitochondriopathy, interferonopathy or both?
Alessandra Pennisi, Agnès Rötig, Charles-Joris Roux, Raphaël Lévy, Marco Henneke, Jutta Gärtner, Pelin Teke Kisa, Fatma Ceren Sarioglu, Uluç Yiş, Laura L Konczal, Deepika D Burkardt, Sulin Wu, Pauline Gaignard, Claude Besmond, Laurence Hubert, Marlène Rio, Giulia Barcia, Arnold Munnich, Nathalie Boddaert, Manuel Schiff
J Med Genet, 2022 Feb, PMID: 33199448 DOI: 10.1136/jmedgenet-2020-107367
– Subclinical maculopathy and retinopathy in transcobalamin deficiency: a 10-year follow-up.
Florence Rigaudière, Hala Nasser, Eliane Delouvrier, Paolo Milani, Manuel Schiff
Doc Ophthalmol, 2022 Feb, PMID: 34491492 DOI: 10.1007/s10633-021-09849-5
– FDX2 and ISCU Gene Variations Lead to Rhabdomyolysis With Distinct Severity and Iron Regulation.
Sebastian Montealegre, Elise Lebigot, Hugo Debruge, Norma Romero, Bénédicte Héron, Pauline Gaignard, Antoine Legendre, Apolline Imbard, Stéphanie Gobin, Emmanuelle Lacène, Patrick Nusbaum, Arnaud Hubas, Isabelle Desguerre, Aude Servais, Pascal Laforêt, Peter van Endert, François Jérome Authier, Cyril Gitiaux, Pascale de Lonlay
Neurol Genet 2022 Jan 19, PMID: 35079622 PMCID: PMC8771665 DOI: 10.1212/NXG.0000000000000648
– Acid Sphingomyelinase Deficiency: Sharing Experience of Disease Monitoring and Severity in France.
Wladimir Mauhin, Raphaël Borie, Florence Dalbies, Claire Douillard, Nathalie Guffon, Christian Lavigne, Olivier Lidove, Anaïs Brassier
J Clin Med, 2022 Feb 10, PMID: 35207195 PMCID: PMC8877564 DOI: 10.3390/jcm11040920
– What are the clues for an inherited metabolic disorder in Reye syndrome? A single Centre study of 58 children.
Violette Goetz, David Dawei Yang, Florence Lacaille, Michele Pelosi, François Angoulvant, Anais Brassier, Jean-Baptiste Arnoux, Manuel Schiff, Claire Heilbronner, Elodie Salvador, Dominique Debray, Mehdi Oualha, Sylvain Renolleau, Muriel Girard, Pascale de Lonlay
Mol Genet Metab, 2022 Apr, PMID: 35221207 DOI: 10.1016/j.ymgme.2022.02.001
– Influence of early identification and therapy on long-term outcomes in early-onset MTHFR deficiency.
Mathilde Yverneau, Stéphanie Leroux, Apolline Imbard, Florian Gleich, Alina Arion, Caroline Moreau, Marie-Cécile Nassogne, Marie Szymanowski, Marine Tardieu, Guy Touati, María Bueno, Kimberly A. Chapman, Yin-Hsiu Chien, Martina Huemer, Pavel Ješina, Mirian C. H. Janssen, Stefan Kölker, Viktor Kožich, Christian Lavigne, Allan Meldgaard Lund, Fanny Mochel, Andrew Morris, Mónica Ruiz Pons, Gloria Liliana Porras-Hurtado, Jean-François Benoist, Léna Damaj, Manuel Schiff, E-HOD Consortium
J Inherit Metab Dis, 2022 Jul, PMID: 35460084 DOI: 10.1002/jimd.12504
– Covid-19: Possible trigger of SLC13A3 reversible leukoencephalopathy relapse?
Apolline Imbard, Julie Pernet, Clément Tarrano, Denis Lacroix, Monique Elmaleh-Bergès, Manuel Schiff
Mol Genet Metab, 2022 Jun, PMID: 35527102 DOI: 10.1016/j.ymgme.2022.04.007
– Intravenous administration of a branched-chain amino-acid-free solution in children and adults with acute decompensation of maple syrup urine disease: a prospective multicentre observational study.
Jean-Meidi Alili, Marie-Pierre Berleur, Marie-Caroline Husson, Karine Mention, Manuel Schiff, Jean-Baptiste Arnoux, Anaïs Brassier, Anne-Sophie Guemman, Coraline Grisel, Sandrine Dubois, Marie-Thérèse Abi-Wardé, Christine Broissand, Aude Servais, Myriam Dao & Pascale de Lonlay
Orphanet J Rare Dis, 2022 May 16, PMID: 35578286 PMCID: PMC9112564 DOI: 10.1186/s13023-022-02353-2
– Neonatal gene therapy achieves sustained disease rescue of maple syrup urine disease in mice.
Clément Pontoizeau, Marcelo Simon-Sola, Clovis Gaborit, Vincent Nguyen, Irina Rotaru, Nolan Tual, Pasqualina Colella, Muriel Girard, Maria-Grazia Biferi, Jean-Baptiste Arnoux, Agnès Rötig, Chris Ottolenghi, Pascale de Lonlay, Federico Mingozzi, Marina Cavazzana, Manuel Schiff
Nat Commun, 2022 Jun 7, PMID: 35672312 PMCID: PMC9174284 DOI: 10.1038/s41467-022-30880-w
– Efficacy and pharmacokinetics of betaine in CBS and cblC deficiencies: a cross-over randomized controlled trial.
Apolline Imbard, Artemis Toumazi, Sophie Magréault, Nuria Garcia-Segarra, Dimitri Schlemmer, Florentia Kaguelidou, Isabelle Perronneau, Jérémie Haignere, Hélène Ogier de Baulny, Alice Kuster, François Feillet, Corinne Alberti, Sophie Guilmin-Crépon, Jean-François Benoist & Manuel Schiff
Orphanet J Rare Dis, 2022 Nov 14, PMID: 36376887 PMCID: PMC9664596 DOI: 10.1186/s13023-022-02567-4
– Initial presentation, management and follow-up data of 33 treated patients with hereditary tyrosinemia type 1 in the absence of newborn screening.
Hela Hajji, Apolline Imbard, Anne Spraul, Ludmia Taibi, Valérie Barbier, Dalila Habes, Anaïs Brassier, Jean-Baptiste Arnoux, Juliette Bouchereau, Samia Pichard, Samira Sissaoui, Florence Lacaille, Muriel Girard, Dominique Debray, Pascale de Lonlay, Manuel Schiff
Mol Genet Metab Rep, 2022 Nov 8, PMID: 36393896 PMCID: PMC9649935 DOI: 10.1016/j.ymgmr.2022.100933
– Branched-Chain Amino Acids and Insulin Resistance, from Protein Supply to Diet-Induced Obesity.
Jean-Pascal De Bandt, Xavier Coumoul, Robert Barouki
Nutrients, 2022 Dec 23, PMID: 36615726 PMCID: PMC9824001 DOI: 10.3390/nu15010068
2021
– Clinical and biological characterization of 20 patients with TANGO2 deficiency indicates novel triggers of metabolic crises and no primary energetic defect.
Claire-Marine Bérat, Sebastian Montealegre, Arnaud Wiedemann, Malou Le Corronc Nuzum, Amélie Blondel, Hugo Debruge, Aline Cano, Brigitte Chabrol et al.
J Inherit Metab Dis, 2021 Mar, PMID: 32929747 DOI: 10.1002/jimd.12314
– Long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency and progressive retinopathy: one case report followed by ERGs, VEPs, EOG over a 17-year period.
Florence Rigaudière, Eliane Delouvrier, Jean-François Le Gargasson, Paolo Milani, Hélène Ogier de Baulny, Manuel Schiff
Doc Ophthalmol, 2021 Jun, PMID: 33392894 DOI: 10.1007/s10633-020-09802-y
– Real-world management of maple syrup urine disease (MSUD) metabolic decompensations with branched chain amino acid-free formulas in France and Germany: A retrospective observational study.
Pascale de Lonlay, Roland Posset, Ulrike Mütze, Karine Mention, Delphine Lamireau, Manuel Schiff, Aude Servais, Jean Baptiste Arnoux, Anaïs Brassier, Myriam Dao et al.
JIMD Rep, 2021 Mar 6, PMID: 33977036 PMCID: PMC8100389 DOI: 10.1002/jmd2.12207
– Quantitative analysis of the natural history of prolidase deficiency: description of 17 families and systematic review of published cases.
Francis Rossignol, Marvid S Duarte Moreno, Jean-François Benoist, Manfred Boehm, Emmanuelle Bourrat, Aline Cano, Brigitte Chabrol, Claudine Cosson, José Luís Dapena Díaz et al.
Genet Med, 2021 Sep, PMID: 34040193 PMCID: PMC8463480 DOI: 10.1038/s41436-021-01200-2
– Compromised mitochondrial quality control triggers lipin1-related rhabdomyolysis.
Yamina Hamel, François-Xavier Mauvais, Marine Madrange, Perrine Renard, Corinne Lebreton, Ivan Nemazanyy, Olivier Pellé, Nicolas Goudin, Xiaoyun Tang, Mathieu P Rodero, Caroline Tuchmann-Durand, Patrick Nusbaum, David N Brindley, Peter van Endert, Pascale de Lonlay
Cell Rep Med, 2021 Aug 17, PMID: 34467247 PMCID: PMC8385327 DOI: 10.1016/j.xcrm.2021.100370
– Liver and brain differential expression of one-carbon metabolism genes during ontogenesis.
Apolline Imbard, Leslie Schwendimann, Sophie Lebon, Pierre Gressens, Henk J Blom, Jean-François Benoist
Sci Rep, 2021 Oct 26, PMID: 34702858 PMCID: PMC8548596 DOI: 10.1038/s41598-021-00311-9
– Sebelipase alfa enzyme replacement therapy in Wolman disease: a nationwide cohort with up to ten years of follow-up.
Tanguy Demaret, Florence Lacaille, Camille Wicker, Jean-Baptiste Arnoux, Juliette Bouchereau, Claire Belloche, Cyril Gitiaux, David Grevent, Christine Broissand, Dalila Adjaoud, Marie-Thérèse Abi Warde, Dominique Plantaz, Soumeya Bekri, Pascale de Lonlay, Anaïs Brassier
Orphanet J Rare Dis, 2021 Dec 14, PMID: 34906190 PMCID: PMC8670257 DOI: 10.1186/s13023-021-02134-3
– Bi-allelic loss-of-function OBSCN variants predispose individuals to severe recurrent rhabdomyolysis.
Macarena Cabrera-Serrano, Laure Caccavelli, Marco Savarese, Anna Vihola, Manu Jokela, Mridul Johari, Thierry Capiod, Marine Madrange, Enrico Bugiardini, Stefen Brady et al.
Brain, 2021 Dec 27, PMID: 34957489 DOI: 10.1093/brain/awab484
– A Review of Multiple Mitochondrial Dysfunction Syndromes, Syndromes Associated with Defective Fe-S Protein Maturation.
Elise Lebigot, Manuel Schiff, Marie-Pierre Golinelli-Cohen
Biomedicines, 2021 Aug 10, PMID: 34440194 PMCID: PMC8393393 DOI: 10.3390/biomedicines9080989
– OTC deficiency in females: Phenotype-genotype correlation based on a 130-family cohort.
Stephanie Gobin-Limballe, Chris Ottolenghi, Fabien Reyal, Jean-Baptiste Arnoux, Maryse Magen, Marie Simon, Anaïs Brassier, Fabienne Jabot-Hanin, Pascale De Lonlay et al.
J Inherit Metab Dis, 2021 Sep, PMID: 34014569 DOI: 10.1002/jimd.12404
– An Unusual Peak in a Common Clinical Presentation.
Bénédicte Sudrié-Arnaud, Sarah Snanoudj, Apolline Imbard, Ivana Dabaj, Abdellah Tebani
Clin Chem, 2021 Apr 29, PMID: 33928370 DOI: 10.1093/clinchem/hvab012
– Adolescent-Onset and Adult-Onset Vitamin-Responsive Neurogenetic Diseases: A Review.
Daniele Mandia, Natalia Shor, Jean-François Benoist, Yann Nadjar
JAMA Neurol, 2021 Apr 1, PMID: 33427863 DOI: 10.1001/jamaneurol.2020.4911
2020
– Long-term outcome of methylmalonic aciduria after kidney, liver, or combined liver-kidney transplantation: The French experience.
Brassier A, Krug P, Lacaille F, Pontoizeau C, Krid S, Sissaoui S, Servais A, Arnoux JB, Legendre C, Charbit M, Scemla A, Francoz C, Benoist JF, Schiff M, Mochel F, Touati G, Broué P, Cano A, Tardieu M, Querciagrossa S, Grévent D, Boyer O, Dupic L, Oualha M, Girard M, Aigrain Y, Debray D, Capito C, Ottolenghi C, Salomon R, Chardot C, de Lonlay P. J Inherit Metab Dis. 2020 Mar;43(2):234-243. doi: 10.1002/jimd.12174. Epub 2020 Feb 11. PMID: 31525265
– Infectious and digestive complications in glycogen storage disease type Ib: Study of a French cohort.
Wicker C, Roda C, Perry A, Arnoux JB, Brassier A, Castelle M, Servais A, Donadieu J, Bouchereau J, Pigneur B, Labrune P, Ruemmele FM, de Lonlay P. Mol Genet Metab Rep. 2020 Apr 9;23:100581. doi: 10.1016/j.ymgmr.2020.100581. eCollection 2020 Jun. PMID: 32300528 Free PMC article.
– Neonatal factors related to survival and intellectual and developmental outcome of patients with early-onset urea cycle disorders.
Pontoizeau C, Roda C, Arnoux JB, Vignolo-Diard P, Brassier A, Habarou F, Barbier V, Grisel C, Abi-Warde MT, Boddaert N, Kuster A, Servais A, Kaminska A, Hennequin C, Dupic L, Lesage F, Touati G, Valayannopoulos V, Chadefaux-Vekemans B, Oualha M, Eisermann M, Ottolenghi C, de Lonlay P. Mol Genet Metab. 2020 Jun;130(2):110-117. doi: 10.1016/j.ymgme.2020.03.003. Epub 2020 Mar 19. PMID: 32273051
– Clinical and biological characterization of 20 patients with TANGO2 deficiency indicates novel triggers of metabolic crises and no primary energetic defect.
Bérat CM, Montealegre S, Wiedemann A, Nuzum MLC, Blondel A, Debruge H, Cano A, Chabrol B, Hoebeke C, Polak M, Stoupa A, Feillet F, Torre S, Boddaert N, Bruel H, Barth M, Damaj L, Abi-Wardé MT, Afenjar A, Benoist JF, Madrange M, Caccavelli L, Renard P, Hubas A, Nusbaum P, Pontoizeau C, Gobin S, van Endert P, Ottolenghi C, Maltret A, de Lonlay P. J Inherit Metab Dis. 2020 Sep 14. doi: 10.1002/jimd.12314. Online ahead of print. PMID: 32929747
– Administration of gamma-hydroxybutyrate instead of beta-hydroxybutyrate to a liver transplant recipient suffering from propionic acidemia and cardiomyopathy: A case report on a medication prescribing error.
Tuchmann-Durand C, Thevenet E, Moulin F, Lesage F, Bouchereau J, Oualha M, Khraiche D, Brassier A, Wicker C, Gobin-Limballe S, Arnoux JB, Lacaille F, Wicart C, Coat B, Schlattler J, Cisternino S, Renolleau S, Secretan PH, De Lonlay P. JIMD Rep. 2020 Jan 3;51(1):25-29. doi: 10.1002/jmd2.12090. eCollection 2020 Jan. PMID: 32071836 Free PMC article.
– Long term outcome of MPI-CDG patients on D-mannose therapy.
Girard M, Douillard C, Debray D, Lacaille F, Schiff M, Vuillaumier-Barrot S, Dupré T, Fabre M, Damaj L, Kuster A, Torre S, Mention K, McLin V, Dobbelaere D, Borgel D, Bauchard E, Seta N, Bruneel A, De Lonlay P. J Inherit Metab Dis. 2020 Jul 17. doi: 10.1002/jimd.12289.
– Long-Term Follow-up of a Child With Putative Remethylation Disorder Who Presented With Severe Anemia as a Neonate.
Zühre Kaya, Manuel Schiff, Jean-François Benoist
J Pediatr Hematol Oncol, 2020 May, PMID: 31789780 DOI: 10.1097/MPH.0000000000001689
– Nocturnal enteral nutrition is therapeutic for growth failure in Fanconi-Bickel syndrome.
Alessandra Pennisi, Bruno Maranda, Jean-François Benoist, Véronique Baudouin, Odile Rigal, Samia Pichard, René Santer, Francesca Romana Lepri, Antonio Novelli, Hélène Ogier de Baulny, Carlo Dionisi-Vici, Manuel Schiff
J Inherit Metab Dis, 2020 May, PMID: 31816104 DOI: 10.1002/jimd.12203
– Administration of gamma-hydroxybutyrate instead of beta-hydroxybutyrate to a liver transplant recipient suffering from propionic acidemia and cardiomyopathy: A case report on a medication prescribing error.
Caroline Tuchmann-Durand,Eloise Thevenet,Florence Moulin,Fabrice Lesage,Juliette Bouchereau,Mehdi Oualha,Diala Khraiche,Anaïs Brassier,Camille Wicker,Stéphanie Gobin-Limballe,Jean-Baptiste Arnoux,Florence Lacaille,Clotilde Wicart,Bruno Coat,Joel Schlattler,Salvatore Cisternino,Sylvain Renolleau,Philippe-Henri Secretan,Pascale De Lonlay
JIMD Rep, 2020 Jan 3, PMID: 32071836 PMCID: PMC7012734 DOI: 10.1002/jmd2.12090
– Nitrous oxide and vitamin B12 in sickle cell disease: Not a laughing situation.
Camille Desprairies, Apolline Imbard, Bérengère Koehl, Mathie Lorrot, Jean Gaschignard, Julie Sommet, Samia Pichard, Laurent Holvoet, Albert Faye, Malika Benkerrou, Jean-François Benoist, Manuel Schiff
Mol Genet Metab Rep, 2020 Mar 17, PMID: 32195121 PMCID: PMC7078522 DOI: 10.1016/j.ymgmr.2020.100579
– [An acidosis not so basic].
Bertrand Lefrère, Emmanuelle Ecochard-Dugelay, Alexis Mosca, Jean-François Benoist, Jean-Pierre Hugot, Apolline Imbard
Ann Biol Clin (Paris), 2020 Aug 1, PMID: 32753366 DOI: 10.1684/abc.2020.1573
– [Biochemical diagnosis of inherited metabolical diseases: metabolic profiles and difficulties for validating methods].
Jean-François Benoist, Roselyne Garnotel, Cécile Acquaviva Bourdain
Ann Biol Clin (Paris), 2020 Oct 1, PMID: 32933895 DOI: 10.1684/abc.2020.1584
– Inherited Disorders of Lysine Metabolism: A Review.
Juliette Bouchereau, Manuel Schiff
J Nutr, 2020 Oct 1, PMID: 33000154 DOI: 10.1093/jn/nxaa112
– Heterogeneity of PNPT1 neuroimaging: mitochondriopathy, interferonopathy or both?
Alessandra Pennisi, Agnès Rötig, Charles-Joris Roux, Raphaël Lévy, Marco Henneke, Jutta Gärtner, Pelin Teke Kisa, Fatma Ceren Sarioglu, Uluç Yiş, Laura L Konczal, Deepika D Burkardt, Sulin Wu, Pauline Gaignard, Claude Besmond, Laurence Hubert, Marlène Rio, Giulia Barcia, Arnold Munnich, Nathalie Boddaert, Manuel Schiff
J Med Genet, 2020 Nov 16, PMID: 33199448 DOI: 10.1136/jmedgenet-2020-107367
2019
– Diagnostic contribution of metabolic workup for neonatal inherited metabolic disorders in the absence of expanded newborn screening
Bower, A ; Imbard, A ; Benoist, JF ; Pichard, S ; Rigal, O ; Baud, O & al.
Sci Rep, 2019 Oct 1, PMID: 31575911 PMCID: PMC6773867 DOI: 10.1038/s41598-019-50518-0
– Clinical, biochemical and genetic characteristics of a cohort of 101 French and Italian patients with HPRT deficiency
Madeo, A ; Di Rocco, M ; Brassier, A ; Bahi-Buisson, N ; De Lonlay, P ; Ceballos-Picot, I.
Mol Genet Metab, 2019 Jun, PMID: 31182398 DOI: 10.1016/j.ymgme.2019.06.001
– Lipin1 deficiency causes sarcoplasmic reticulum stress and chaperone-responsive myopathy
Rashid, T ; Nemazanyy, I ; Paolini, C ; Tatsuta, T ; Crespin, P ; de Villeneuve, D & al.
EMBO J, 2019 Jan 3, PMID: 30420558 PMCID: PMC6315296 DOI: 10.15252/embj.201899576
– Elevated thrombin generation in patients with congenital disorder of glycosylation and combined coagulation factor deficiencies
Pascreau, T ; de la Morena-Barrio, ME ; Lasne, D ; Serrano, M ; Bianchini, E ; Kossorotoff, M & al.
J Thromb Haemost, 2019 Nov, PMID: 31271700 DOI: 10.1111/jth.14559
– International clinical guidelines for the management of phosphomannomutase 2-congenital disorders of glycosylation: Diagnosis, treatment and follow up
Altassan, R ; Péanne, R ; Jaeken, J ; Barone, R ; Bidet, M ; Borgel, D & al.
J Inherit Metab Dis, 2019 Jan, PMID: 30740725 DOI: 10.1002/jimd.12024
2018
– Long-term liver disease in methylmalonic and propionic acidemias
Imbard, A ; Garcia Segarra, N ; Tardieu, M ; Broué, P ; Bouchereau, J ; Pichard, S & al.
Mol Genet Metab, 2018 Apr, PMID: 29433791 DOI: 10.1016/j.ymgme.2018.01.009
2017
– Biallelic Mutations in DNAJC12 Cause Hyperphenylalaninemia, Dystonia, and Intellectual Disability
Anikster, Y ; Haack, TB ; Vilboux, T ; Pode-Shakked, B ; Thöny, B ; Shen, N & al.
Am J Hum Genet, 2017 Feb 2, PMID: 28132689 PMCID: PMC5294665 DOI: 10.1016/j.ajhg.2017.01.002
– Neurocognitive profiles in MSUD school-age patients
Bouchereau, J ; Leduc-Leballeur, J ; Pichard, S ; Imbard, A ; Benoist, JF ; Abi Warde, MT & al.
J Inherit Metab Dis, 2017 May, PMID: 28324240 DOI: 10.1007/s10545-017-0033-7
2016
– Riboflavin-Responsive and -Non-responsive Mutations in FAD Synthase Cause Multiple Acyl-CoA Dehydrogenase and Combined Respiratory-Chain Deficiency
Olsen, RKJ ; Koňaříková, E ; Giancaspero, TA ; Mosegaard, S ; Boczonadi, V ; Mataković, L & al.
Am J Hum Genet, 2016 Jun 2, PMID: 27259049 PMCID: PMC4908180 DOI: 10.1016/j.ajhg.2016.04.006
- Association des familles galactosémiques de France (AFGF)
- Vaincre les maladies lysosomales (VML)
- Association pour la lutte contre l’alcaptonurie (ALCAP)
- Association contre les maladies mitochondriales (AMMI)
- TANGO2 Research Foundation
- Connaître les Syndromes Cérébelleux (CSC)
- Association Francophone des Glycogénoses (AFG)
- Fructos’Amis pour la vie
- Les Feux Follets
- Les enfants du jardin
- Association syndrome de Barth
- Association Noa Luu mon combat
- Lesch-Nyhan action
- Association des Hyperinsulinismes
- Nos anges
- Les p’tits CDG
- No Myolyse
- Nos enfants Menkes
- Association XTRAORDINAIRE
- Ensemble contre la Tyrosinémie
- Alliance Maladies Rares
- AG1-23 Soleil – Acidurie Glutarique de Type 1
- Alliance Sanfilippo
- Association des Patients de la Maladie de Fabry (APMF)
- Association sur le déficit en Glut 1 (ASD GLUT1)
- Les Petits Bourdons
- AFM Téléthon
Consulter l’annuaire des associations de la filière G2M
Regarder la vidéo des associations partenaires de la filière G2M
Télécharger le poster des associations de la filière G2M
2022
- Ryanodine receptor type 3 variants cause acute episodes of rhabdomyolysis related to abnormal calcium homeostasis and impaired autophagy (H de Calbiac, P de Lonlay), SSIEM Annual Symposium 30/08-02/09 2022, Freiburg, Germany; congrès européen et international pour les métaboliciens
- Systemic corticosteroids for the treatment of acute episodes of rhabdomyolysis in lipin-1-deficient patients (P de Lonlay), SSIEM Annual Symposium 30/08-02/09 2022, Freiburg, Germany; congrès européen et international pour les métaboliciens
- Sustained efficacy of neonatal AAV gene therapy for maple syrup urine disease in mice (C Pontoizeau), SSIEM Annual Symposium 30/08-02/09 2022, Freiburg, Germany; congrès européen et international pour les métaboliciens
- Influence of early identification and therapy on long-term outcomes in early-onset MTHFR deficiency (M Schiff), SSIEM Annual Symposium 30/08-02/09 2022, Freiburg, Germany; congrès européen et international pour les métaboliciens
- Rhabdomyolyses aigues – (P de Lonlay) intervention Session Enseignement de la SFMyologie 2022 Toulouse 23/11/2022, congrès pour les médecins neuromusculaires
2021
- E-journée Pompe
18 janvier 2021 | Visioconférence
>> En savoir +
>> inscription
- Sebelipase alfa enzyme replacement therapy in Wolman disease: a nationwide cohort with up to ten years of follow-up; SSEIM, virtual conference (Covid-19); Tanguy Demaret et Anais Brassier; Europe, métaboliciens
- TANGO2 deficiency: guidelines and zebrafish model; Fondation TANGO2 Research, virtual conference; Pascale de Lonlay et Hortense de Calbiac; cliniciens, chercheurs et patients, International
- Programme national du dépistage néonatal : les 7 futures maladies héréditaires du métabolisme; Société Française de Pédiatrie (digitale); Jean-Baptiste Arnoux; pédiatres, National
- Etude prospective Protect : Carbaglu au long cours dans les aciduries organiques; Société Française des Erreurs Innées du Métabolise (SFEIM, juin); Anais Brassier; Métaboliciens, National
- Dépistage néonatal des maladies de surcharge lysosomales : une vaste question aux confins du faisable, de l’utile et de l’éthique : état des lieux du dépistage néonatal, état actuel des options thérapeutiques, avancées en France sur le projet de DNN; SFDN; Anais Brassier; pédiatres et soignants, National
2020
- Rhabdomyolyses, P de Lonlay, Société Française Erreurs Innées du Métabolisme (SFEIM); métabolicien
- « Les perspectives du dépistage des maladies métaboliques : quel impact ?, JB Arnoux, « Symposium de la Société Française de Dépistage Néonatal.
- Déficits en TANGO2, S Montealegre/P de Lonlay; LPIN1, Paris, P Renard/P de Lonlay, Journée recherche filière G2M; métaboliciens, biologistes, chercheurs.
- Symposium industriel (SOBI) international sur tyrosinémie de type 1, M Schiff: co organisateur et 2 com. orales; métaboliciens.
- Médicaments orphelins, P de Lonlay, Alliance Maladies Rares; associations de patients, médecins maladies rares, Cnam, AGEPS, ANSM, DGOS.
2018
8e journée française sur la maladie de Pompe | Institut Imagine | 30.3.2018
> Ce qui s’y est dit est ici
Dépistage néonatal : déficit en MCAD, une sixième maladie détectée dès la naissance
Le Parisien | 14.12.2020
Depuis le 1er décembre, tous les nouveau-nés sont dépistés pour le déficit en acyl-CoA déshydrogénase des acides gras à chaîne moyenne (MCAD). Cela s’ajoute aux cinq autres pathologies déjà recherchées.
> Lire la suite
Coordonnées du CRMR
Hôpital universitaire Necker-Enfants malades
> Maladies métaboliques pédiatriques
149 rue de Sèvres
75743 PARIS Cedex 15
> Livret d’accueil pédiatrique
![]()
À Necker, le centre de référence des maladies héréditaires du métabolisme de l’enfant et de l’adulte (MAMEA) c’est …
* données pour l’année 2022